- By
- November 27, 2025
- Revita Nerve
Diabetic neuropathy presents a major public health problem. It is defined by the symptoms and signs of peripheral nerve dysfunction in diabetic patients, in whom other causes of neuropathy have been excluded. Pathogenetic mechanisms that have been implicated in diabetic neuropathy are: a) increased flux through the polyol pathway, leading to accumulation of sorbitol, a reduction in myo-inositol, and an associated reduced Na+-K+-ATPase activity, and b) endoneurial microvascular damage and hypoxia due to nitric oxide inactivation by increased oxygen free radical activity. Alpha-lipoic acid seems to delay or reverse peripheral diabetic neuropathy through its multiple antioxidant properties. Treatment with alpha-lipoic acid increases reduced glutathione, an important endogenous antioxidant. In clinical trials, 600 mg alpha-lipoic acid has been shown to improve neuropathic deficits. This review focuses on the relationship of alpha-lipoic acid and auto-oxidative glycosylation. It discusses the impact of alpha-lipoic acid on hyperglycemia-induced oxidative stress, and examines the role of alpha-lipoic acid in preventing glycation process and nerve hypoxia.
Keywords: diabetes, alpha-lipoic acid, reactive oxygen species, advanced glycation end products, nuclear factor-kappaB, protein kinase C
Introduction
Diabetic neuropathy is defined by the signs and symptoms of peripheral nerve dysfunction in diabetic patients, in whom other causes of neuropathy have been excluded [1]. Diabetic neuropathy includes a number of different syndromes, depending on the classes of nerve fibers involved [2]. According to the San Antonio Convention, the major groups of neurologic disorders in patients with diabetes mellitus are: 1. subclinical neuropathy defined by abnormalities in electrodiagnostic and quantitative sensory testing, 2. diffuse clinical neuropathy with distal sensorimotor and autonomic syndromes, and 3. focal syndromes [3].
At least 25% of diabetic patients are affected by distal symmetric polyneuropathy, which is a major public health problem, as it is responsible for considerable morbidity and mortality [4–7]. Distal symmetric polyneuropathy is a major contributing factor for diabetic foot ulcer, osteoarthopathy, osteomyelitis, and lower limb amputation. The latter is fifteen times higher in diabetic patients than in the general population [5, 8]. Neuropathic pain affects approximately 16% of diabetic patients [9]. This subjective symptom impairs quality of life and sleeping as it usually gets worse at night [1, 8]. It is often the major complaint that motivates patients to seek health care [10]. However, treatment of painful diabetic symmetric polyneuropathy is still a challenge for the physician [11].
Treatment of diabetic neuropathy is based on: 1. aiming at near-normoglycemia, 2. pathogenetically oriented therapy, 3. symptomatic therapy, and 4. avoidance of risk factors [8]. Near-normoglycemia is generally accepted as the first approach towards preventing diabetic neuropathy [12, 13]. As normoglycemia is difficult to achieve, additional treatment of painful symptoms is frequently required [14]. Pathogenetically oriented therapy may delay, stop, or reverse the progression of neuropathy and may alleviate pain. Whilst symptomatic therapy does not influence the course leading to neuropathy, it may alleviate painful symptoms [15].
Antidepressants (SSRIs and tricyclic), opioids (e.g. controlled-release oxycodone), and older anticonvulsants (e.g. carbamazepine) all seem to alleviate pain, but have several adverse side effects [16]. Newer anticonvulsants such as gabapentin and pregabalin have a high affinity binding to α2-δ subunit of voltage-activated calcium channels. They combat painful diabetic neuropathy, partly via calcium channel modulation in the pathogenesis of diabetic neuropathy [16, 17].
Pathogenesis of diabetic neuropathy
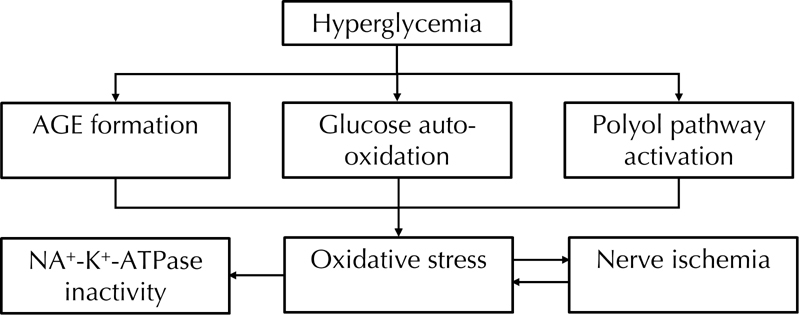
The pathogenesis of diabetic neuropathy is complicated. The following mechanisms seem to be involved: 1. increased flux through the polyol pathway, mediated by aldose reductase and sorbitol dehydrogenase, leading to accumulation of sorbitol and depletion of myo-inositol. The latter reduction is associated with reduced Na+-K+-ATPase activity [8]. 2. Endoneurial microvascular damage and hypoxia due to nitric oxide inactivation [18]. 3. Accumulation of advanced glycation end products (AGEs) that exert their damaging effects by binding to specific receptors on the surface of neurons. Binding of AGEs to their receptors causes oxidative stress and activates nuclear factor-κB (NF-κB). There is increasing evidence that the diverse agents able to activate NF-κB elevate levels of reactive oxygen species (ROS). Also, chemically distinct antioxidants and overexpression of antioxidant enzymes can inhibit NF-κB activation [8, 18–20]. 4. Increased nerve lipid peroxidation in vivo. The most reliable index of increased oxidative stress is reduction in GSH [18]. 5. Activation of protein kinase C (PKC) by increased release of intracellular diacylglycerol (DAG) due to glycolysis. Hyperglycemia activates PKC, especially its βII isoform through increased de novo synthesis of DAG. The increased activity of PKCβ may impair endoneurial blood flow. Recently, hyperglycemia has been associated with activation of PKC and increase in Nav1.7 tetrodotoxin-sensitive voltage-gated sodium channel isoform; both of which play a critical role in the perception of pain [21–24]. 6. Alterations in mitogen-activated protein kinases (MAPKs) result in a signaling cascade involved in the pathogenesis of peripheral diabetic neuropathy [25]. 7. Abnormal Ca2+ homeostasis and signaling [26].
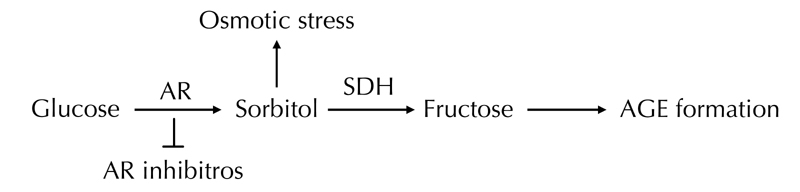
As mentioned above, hyperglycemia-induced ischemic and auto-oxidative lipid peroxidation is suggested to cause diabetic neuropathy (Figure 1). Streptozotocin-induced experimental diabetic neuropathy in rats is associated with a nerve blood flow deficit of 50% [18]. Endoneurial hypoxia is secondary to a reduction in nerve blood flow and increased endoneurial vascular resistance. Hyperglycemia acts via a reduction in nitric oxide resulting in impaired microvascular tone, reduced nerve blood flow, and endoneurial hypoxia [27, 28]. Hypoxic nerve can continue to function on glucose alone under anoxic circumstances via anaerobic glycolysis. A detrimental effect is hyperactivity of the polyol pathway resulting in an increase in sorbitol and a reduction in myo-inositol [27–30]. Nevertheless, sorbitol per se is non-toxic and it seems likely that mechanisms other than nerve sorbitol accumulation cause neuropathy. The reduction in myo-inositol has been linked to neuropathy via reduction in Na+-K+-ATPase activity [31–37]. The mechanism of myo-inositol reduction by hyperactivity of the polyol pathway is not completely known, but hyperglycemia could reduce nerve myo-inositol by competitive inhibition of peripheral nerve myo-inositol uptake. Osmolyte and non-osmolyte mechanisms could be involved [38, 39].
The effectiveness and tolerability of aldose reductase inhibitors and protein kinase C inhibitors are currently being investigated [40]. Besides, acetyl-L-carnitine is deficient in diabetes. Substitution with acetyl-L-carnitine corrects perturbations of neural Na+-K+-ATPase, myo-inositol, and nitric oxide. It also improves nerve fiber regeneration and alleviates symptoms, particularly pain in patients with established diabetic neuropathy [41–43]. Alpha-lipoic acid seems to normalize endoneurial Na+-K+-ATPase activity in experimental diabetic nerves [44]. As observed in retinal cells, the improved Na+-K+-ATPase activity could improve myo-inositol uptake by the Na+–myo-inositol co-transporter [34–37]. Alpha-lipoic acid has an effect on glucose uptake, thereby increasing polyol pathway activity [44]. It is known to increase Krebs cycle activity, too [45]. The effects of alpha-lipoic acid on glucose uptake and polyol metabolites, as well as the ability of alpha-lipoic acid to increase pyruvate dehydrogenase and α-ketoglutarate activity in a number of non-neural tissues, suggest that the effects of alpha-lipoic acid on the polyol pathway and the Krebs cycle are worth further exploration [44, 45].
Oxidative stress and alpha-lipoic acid
Hyperglycemia-induced oxidative stress induces programmed cell death of nerves, which contributes to the pathology of diabetic neuropathy [46, 47]. A study found reduced frequency of apoptosis in diabetic animals that were treated with the antioxidant taurine [48]. Another study reported increased frequency of programmed cell death in cultured dorsal root ganglia when glucose was added [49]. The role of oxidative stress in nerve damage has been extensively studied in experimental diabetes and in diabetic subjects [50–55]. Motor nerve and sensory nerve conduction velocities are the principal endpoints in studying the therapeutic effectiveness of alpha-lipoic acid on nerve function. Alpha-lipoic acid has been shown to improve motor nerve conduction velocity in experimental diabetic neuropathy and protect peripheral nerves from ischemia in rats [54, 55].
Treatment with alpha-lipoic acid increases reduced glutathione (GSH) in vivo and in vitro [56–59]. GSH is an important endogenous antioxidant. Together with lipoic acid it seem to play a major role in redox-dependent mechanisms of various cellular targets [18, 59–61]. Alpha-lipoic acid is a powerful lipophilic free radical scavenger of peripheral nerve both in vitro and in vivo [62, 63]. As diabetes has been associated with increased production and/or decreased clearance of ROS, oxidative stress has been suggested to contribute to defective nerve blood supply and endoneurial oxidative damage [64, 65]. The increased availability of glucose in diabetes induces enhanced production of AGEs. This process is defined as auto-oxidative glycosylation and is considered the major cause of increased ROS production among diabetic subjects [66]. The increased availability of glucose leads to glycation of antioxidant enzymes [67–70]. Therefore, the process of glucose auto-oxidation might be responsible for enhanced ROS production and for decreased availability or activity of antioxidant enzymes [71, 72]. Furthermore, fructose, which is increased due to the activation of the polyol pathway, leads to the formation of AGE precursors [73] (Figure 2). Alpha-lipoic acid has additional actions such as stimulating nerve growth factor and promoting fiber regeneration [74, 75].
History of alpha-lipoic acid
R-alpha-lipoic acid (1, 2-dithiolane-3-pentanoic acid) was discovered in 1937 by Snell et al., who found that certain bacteria needed a compound from potato extract for growth [76]. In 1951, the so-called potato-growth factor was isolated by Reed and colleagues, and lipoic acid was discovered as a molecule that assists in acyl-group transfer and as a co-enzyme in the Krebs cycle [77, 78]. In the 1980s, alpha-lipoic acid was recognized as a powerful antioxidant. It is the only fat- and water-soluble antioxidant. It is produced by animals and humans [79], and can be found in liver, skeletal muscle, potatoes, and broccoli [80, 81]. Nutritional supplements of alpha-lipoic acid are typically comprised either of R-alpha-lipoic acid alone or a racemic mixture of R-alpha-lipoic acid and S-alpha-lipoic acid [79].
Alpha-lipoic acid in clinical practice
Diabetic patients with neuropathy treated with alpha-lipoic acid 600 mg i.v. daily for three weeks, have reduced pain, paresthesias, and numbness [14, 80, 81]. According to a recent meta-analysis comprising 1,258 patients, the same treatment ameliorated neuropathic symptoms and deficits after three weeks [81]. Acute infusion of alpha-lipoic acid improved nitric oxide-mediated endothelium-dependent vasodilation in diabetic patients, and improved microcirculation in patients with diabetic polyneuropathy [60–65, 81–89].
Oral treatment with alpha-lipoic acid for five weeks improved neuropathic symptoms and deficits in 187 patients with diabetic symmetrical polyneuropathy. This is an encouraging finding as deficits are major risk factors in the development of neuropathic foot ulcer [10, 90–92]. An oral dose of 600 mg once daily seems to provide the optimum risk-to-benefit ratio in the SYDNEY 2 trial [10]. The adverse effects (mainly nausea) with the 1,200 mg dose daily occurred in 21% of patients, somewhat higher than that observed in the ALADIN I (15%) and ALADIN II study (7%), with the same dose of alpha-lipoic acid [10, 93, 94]. In the seven-month ALADIN III trial, 509 subjects received either 600 mg of alpha-lipoic acid or placebo. While no significant difference was noted in subjective symptom evaluation among the groups, treatment with alpha-lipoic acid was associated with improved nerve function [95].
In the ISLAND Study, 300 mg of alpha-lipoic acid was applied as monotherapy and in combination with 150 mg imbesartan daily. There was a significant increase in endothelium-dependent flow-mediated vasodilation of the brachial artery, by 44% and 75% respectively, compared with placebo treatment after four weeks. This effect was accompanied by reductions in plasma levels of interleukin-6 and plasminogen activator-1, suggesting that alpha-lipoic acid may improve endothelial dysfunction via anti-inflammatory and antithrombotic mechanisms [96]. These anti-inflammatory and antithrombotic properties have previously been observed in streptozotocin-diabetic rats by significant decreases in fibrinogen factor VII and von Willebrand factor (vWF) after treatment with alpha-lipoic acid [97].
Alpha-lipoic acid has been shown to downregulate the expression of cell-adhesion molecules ICAM-1 and VCAM-1 in a dose-dependent manner [98]. These observations might be of preventive and/or therapeutic benefit in arteriosclerosis and other inflammatory disorders [18]. Clinical and postmarketing surveillance studies have revealed a highly favorable safety profile of the drug [99]. Nevertheless, further studies are necessary to assess the neurophysiological and clinical properties of alpha-lipoic acid.
Conclusions
Distal symmetric polyneuropathy is a major public health problem causing substantial morbidity and mortality among diabetic patients. Its pathogenesis remains complicated. Increased flux through the polyol pathway, which leads to accumulation of sorbitol, depletion of myo-inositol, endoneurial microvascular damage, and hypoxia seem to be the underlying pathogenetic mechanisms. The latter is due to nitric oxide inactivation and accumulation of AGEs that exert their damaging effects by binding to specific receptors on the surface of neurons, leading to activation of NF-κB.
The role of oxidative stress in nerve damage has been extensively studied in experimental and clinical diabetes. Alpha-lipoic acid has been shown to improve motor-nerve conduction velocity in experimental diabetic neuropathy, and to protect peripheral nerves from ischemia in rats.
Treatment with alpha-lipoic acid increases GSH in vivo and in vitro. GSH is an important endogenous antioxidant, and together with lipoic acid, it seems to play a predominant role in the redox-dependent mechanisms of various cellular targets. As diabetes has been associated with increased production and/or decreased clearance of ROS, oxidative stress has been suggested to contribute to defective nerve blood supply and endoneurial oxidative damage.
Recently, trials have been conducted with neuropathic diabetes patients who received 600 mg alpha-lipoic acid. The treatment reduced pain, paresthesias, and numbness. Further studies are needed to determine the effectiveness of alpha-lipoic acid in reducing pain and/or preventing the progression of diabetic neuropathy.
Disclosures: The authors report no conflict of interests.
1 Comment
https://shorturl.fm/w2av0